





Abstract
Increased mitochondrial reactive oxygen species (ROS), particularly superoxide, have been suggested to mediate hypoxic pulmonary vasoconstriction (HPV), chronic hypoxia-induced pulmonary hypertension and right ventricular remodelling.
We determined ROS in acute and chronic hypoxia, and investigated the effect of the mitochondria-targeted antioxidant MitoQ under these conditions.
The effect of MitoQ or its inactive carrier substance, decyltriphenylphosphonium, on acute HPV (1% O2 for 10 min) was investigated in isolated blood-free perfused mouse lungs. Mice exposed to chronic hypoxia (10% O2 for 4 weeks) or after banding of the main pulmonary artery were treated with MitoQ or decyltriphenylphosphonium (50 mg·kg−1·day−1).
Total cellular superoxide and mitochondrial ROS levels were increased in pulmonary artery smooth muscle cells but decreased in pulmonary fibroblasts in acute hypoxia. MitoQ significantly inhibited HPV and acute hypoxia-induced rise in superoxide concentration. ROS was decreased in pulmonary artery smooth muscle cells, while it increased in the right ventricle after chronic hypoxia. Correspondingly, MitoQ did not affect the development of chronic hypoxia-induced pulmonary hypertension but attenuated right ventricular remodelling after chronic hypoxia as well as after pulmonary arterial banding.
Increased mitochondrial ROS of pulmonary artery smooth muscle cells mediate acute HPV, but not chronic hypoxia-induced pulmonary hypertension. MitoQ may be beneficial under conditions of exaggerated acute HPV.
Abstract
The mitochondria-targeted antioxidant MitoQ attenuates acute hypoxic pulmonary vasoconstriction and RV remodelling, but not chronic hypoxia-induced PH http://ow.ly/PCCU30hSJKK
Introduction
Hypoxic pulmonary vasoconstriction (HPV) is a reaction of the pre-capillary pulmonary vessels to alveolar hypoxia and serves to maintain ventilation–perfusion matching, thereby optimising the oxygenation of blood [1]. In contrast, generalised chronic hypoxia (e.g. in chronic obstructive pulmonary disease) leads to the development of pulmonary hypertension, which is a progressive disorder characterised by pulmonary vascular remodelling, ultimately resulting in right heart failure [1]. In addition to vascular remodelling, generalised HPV contributes to the increased vascular resistance in chronic hypoxia-induced pulmonary hypertension.
Pulmonary arterial smooth muscle cells (PASMCs) are key players in these responses, as they react to acute hypoxia with contraction and to chronic hypoxia with proliferation, even when isolated [2, 3]. Mitochondrial reactive oxygen species (ROS) have been suggested to play a crucial role in both processes by interaction with protein kinases, phospholipases and ion channels inducing intracellular calcium release or by stabilisation of transcription factors [1, 4]. Additionally, ROS may be involved in the development of right ventricular remodelling [5]. However, it remains unclear if ROS are increased or decreased during acute and chronic hypoxia in the pulmonary vasculature and right ventricle, and which species (superoxide or hydrogen peroxide (H2O2)) trigger these responses [1, 3, 6]. Application of the unspecific thiol compound N-acetylcysteine that attenuates many ROS processes could inhibit hypoxia-induced pulmonary hypertension [7]. Overexpression of the mitochondrial superoxide dismutase 2 (SOD2) increased and overexpression of mitochondria-targeted catalase decreased pulmonary vascular remodelling, suggesting that increased mitochondrial H2O2 contributes to hypoxia-induced pulmonary hypertension [8].
MitoQ is an orally available mitochondria-targeted antioxidant, consisting of a ubiquinone moiety linked to a triphenylphosphonium (TPP+) molecule [9]. The lipophilic TPP+ cation allows MitoQ to pass through the phospholipid bilayers and to accumulate within the mitochondrial inner membrane driven by the mitochondrial membrane potential [9]. The positively charged residue (TPP+) of MitoQ is adsorbed to the matrix surface, while the hydrophobic end (ubiquinone) is inserted into the hydrophobic core of the mitochondrial inner membrane [10]. The active antioxidative form of MitoQ, ubiquinol, is oxidised by ROS to the inactive form, ubiquinone, which is continually recycled by complex II of the respiratory chain to its active ubiquinol form [9]. MitoQ is an effective antioxidant against lipid peroxidation, peroxynitrite, the hydroperoxyl radical and superoxide, although its reactivity with H2O2 is negligible [10–12]. However, under specific conditions, particularly in the non-membrane-bound form, MitoQ may also increase superoxide generation [9, 13], which probably only occurs in vitro [9]. MitoQ has been demonstrated to be protective against several ROS-mediated pathologies, including cardiovascular diseases [14] and sepsis [15] in animal models and in humans [16]. As superoxide and H2O2 are suggested to regulate the development of chronic hypoxia-induced pulmonary hypertension, MitoQ offers the possibility to specifically target mitochondrial ROS and oxidative damage therapeutically.
We thus aimed to elucidate the role of mitochondria-derived ROS in HPV, chronic hypoxia-induced pulmonary hypertension and right ventricular remodelling. We hypothesised that the mitochondria-targeted antioxidant MitoQ is useful 1) to investigate the role of mitochondrial ROS in these process and 2) as a possible therapeutic tool.
Materials and methods
Animal experiments and reagents
Animal experiments were approved by the governmental authorities. C57BL/6J mice of either sex were studied. All reagents were from Sigma Aldrich (St Louis, MO, USA), unless stated otherwise. MitoQ and decyl-TPP+ were provided by Antipodean Pharmaceuticals (Auckland, New Zealand).
Isolated blood-free perfused and ventilated mouse lung
Lungs of mice were isolated, ventilated and blood-free perfused as described previously [17].
Chronic hypoxic exposure, pulmonary arterial banding and treatment with MitoQ
For induction of chronic hypoxia-induced pulmonary hypertension, mice were kept under normobaric hypoxia (10% O2) for 28 days [18]. Banding of the main pulmonary artery (pulmonary arterial banding (PAB)) was performed as described previously [19]. During chronic hypoxic incubation and after PAB, mice were treated with 50 mg·kg−1·day−1 MitoQ or TPP+ dissolved in H2O by gavage [9].
Development of pulmonary hypertension was determined by in vivo haemodynamics and histological analysis [18]. Heart function was measured by transthoracic echocardiography [20].
Isolated PASMCs and lung fibroblasts
PASMCs and lung fibroblasts were isolated from pre-capillary pulmonary arteries as described previously [21, 22].
Measurement of ROS release by electron spin resonance spectroscopy and fluorescence approaches
Intracellular and extracellular concentrations of ROS and reactive nitrogen species (RNS) were determined in PASMCs and tissues by a Bruker electron spin resonance (ESR) spectrometer (EMXmicro; Bruker Biospin, Rheinstetten, Germany), using the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH; 0.5 mM) [23, 24]. The superoxide portion of the CMH signal was determined by subtracting the ESR signal of the samples incubated with 50 U·mL−1 poly(ethylene glycol)-conjugated SOD (pSOD) and CMH from the signal of the CMH-only samples.
Mitochondrial ROS were measured by the fluorescent dye MitoSOX. The protein-based fluorescent sensor HyPercyto was used to detect cytosolic H2O2 concentration [25].
MitoQ concentration in the lung and heart
MitoQ concentration was measured by reverse-phase liquid chromatography and tandem mass spectrometry [26].
Proliferation assay
Proliferation of PASMCs was evaluated by determination of the ratio of proliferating cells labelled by 5-ethynyluridine to the total cell number labelled by Hoechst staining [18].
Additional materials and methods
Additional materials and methods are available in the supplementary material.
Statistical methods
Values are given as mean±sem. Statistical significance of the data was calculated by the t-test with Welsh's correction in experiments with two experimental groups and by one- or two-way ANOVA in experiments with more than two experimental groups. Two-way ANOVA was used to verify interactions between the factors of treatment (TPP+ versus MitoQ) and exposure (normoxia/hypoxia or PAB/sham). A p-value <0.05 was considered significant.
Results
MitoQ inhibits acute HPV as well as the hypoxia-induced increase of superoxide in PASMCs
The strength of HPV was determined as the increase in pulmonary arterial pressure (ΔPAP) during hypoxic ventilation in isolated mouse lungs. The first hypoxic manoeuvre in the absence of any substance served as baseline (100%). Application of 0.5 µM MitoQ significantly attenuated the hypoxia-induced elevation of PAP (fourth hypoxic manoeuvre) compared with ΔPAP in the absence of MitoQ (first hypoxic manoeuvre) or ΔPAP in the presence of 0.5 µM TPP+ during the fourth hypoxic manoeuvre (figure 1a). In contrast, higher doses of MitoQ or TPP+ (1 µM) reduced acute HPV to a similar extent (fifth hypoxic manoeuvre) compared with the control (figure 1a). Both MitoQ and TPP+ did not alter U46619-induced vasoconstriction (figure 1b), displaying the specificity of the MitoQ effect for HPV. The strength of HPV and U46619-induced pulmonary vasoconstriction was similar in all experimental groups prior to the application of MitoQ or TPP+ (supplementary figure S1a and b).
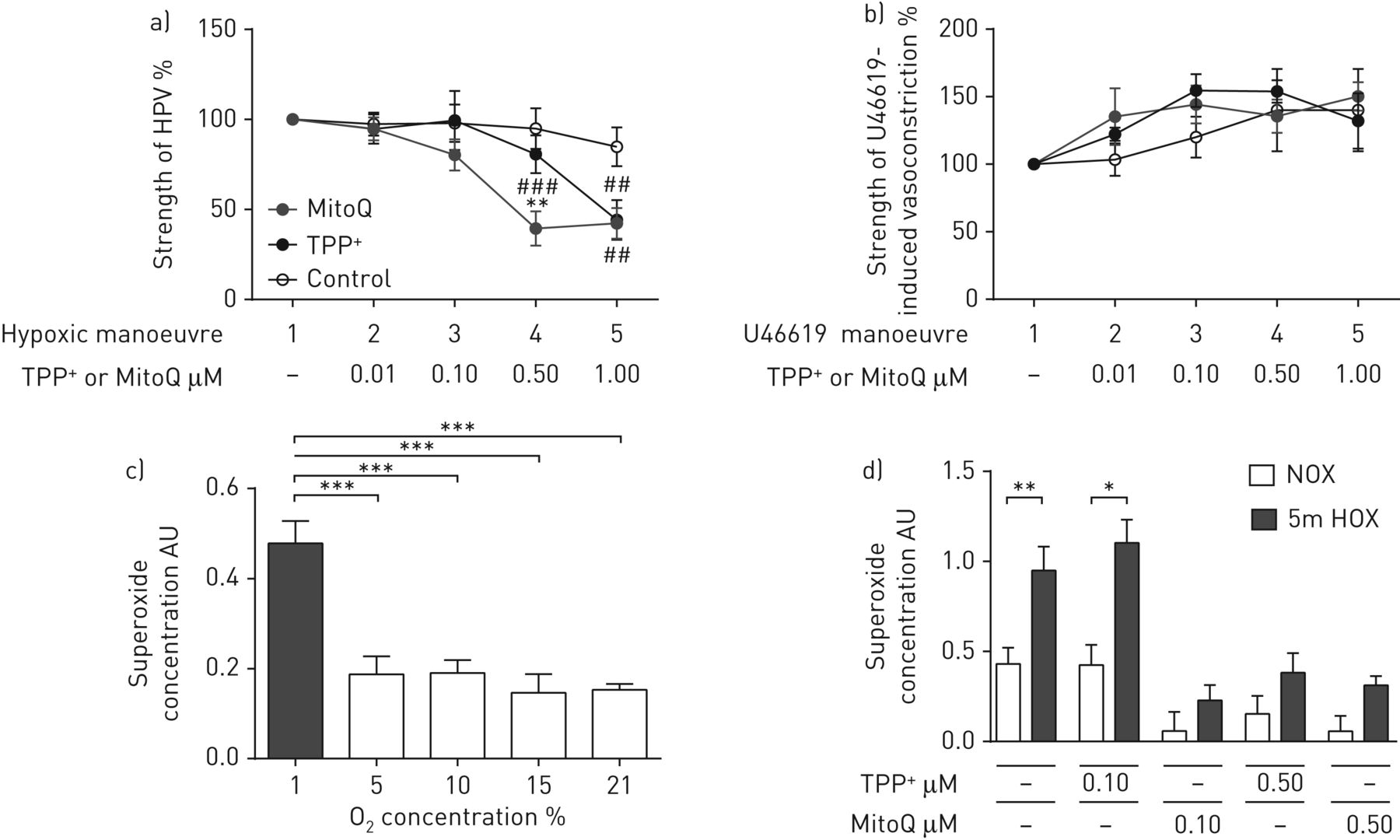
Effect of MitoQ on hypoxic pulmonary vasoconstriction (HPV) in isolated ventilated and perfused mouse lungs and on superoxide concentration in pulmonary artery smooth muscle cells (PASMCs) during acute hypoxia. a, b) Increase of pulmonary arterial pressure in isolated ventilated and perfused mouse lungs a) during acute hypoxic ventilation (1% O2, 5.3% CO2, balanced with N2) or b) after bolus injection of the thromboxane mimetic U46619 in the absence (control) or presence of the mitochondria-targeted antioxidant (MitoQ) or the inactive carrier substance (triphenylphosphonium (TPP+)). n=5–6 isolated lungs in each group. **: p<0.01 compared with the TPP+-treated group; ##: p<0.01; ###: p<0.001 compared with the respective control groups by two-way ANOVA with the Tukey post hoc test. c) Superoxide concentration in PASMCs after 5 min of acute exposure to different O2 concentrations. The superoxide concentration was measured by electron spin resonance spectroscopy as the difference in the 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine signal with or without poly(ethylene glycol)-conjugated superoxide dismutase. AU: arbitrary units. n=3–4 individual cell isolations per group. ***: p<0.001 by one-way ANOVA with the Tukey post hoc test. d) Superoxide concentration in PASMCs after either 5 min of acute hypoxic (1% O2, rest N2 (“5m HOX”)) or normoxic (21% O2 (“NOX”)) incubation in the presence or absence of the mitochondria-targeted antioxidant (MitoQ) or the inactive carrier substance (TPP+). n=5–6 individual cell isolations per group. *: p<0.05; **: p<0.01 by two-way ANOVA with the Tukey post hoc test.
Exposure of PASMCs to 1% O2 but not to 5%, 10% and 15% O2 for 5 min induced an increase of the intra- and extracellular superoxide concentration measured by ESR spectroscopy (figure 1c). 0.1 µM MitoQ specifically inhibited the acute hypoxia-induced increase of superoxide, while no effect of TPP+ at this concentration was detected (figure 1d). At higher concentrations (0.5 µM) both MitoQ and TPP+ caused a significant decrease of superoxide concentration in normoxia and hypoxia.
The values of O2 tension, CO2 tension and pH were monitored during representative experiments (supplementary figure S2a−f).
Exposure to acute hypoxia induces an increase of cytosolic H2O2, mitochondrial ROS and total cellular superoxide specifically in PASMCs
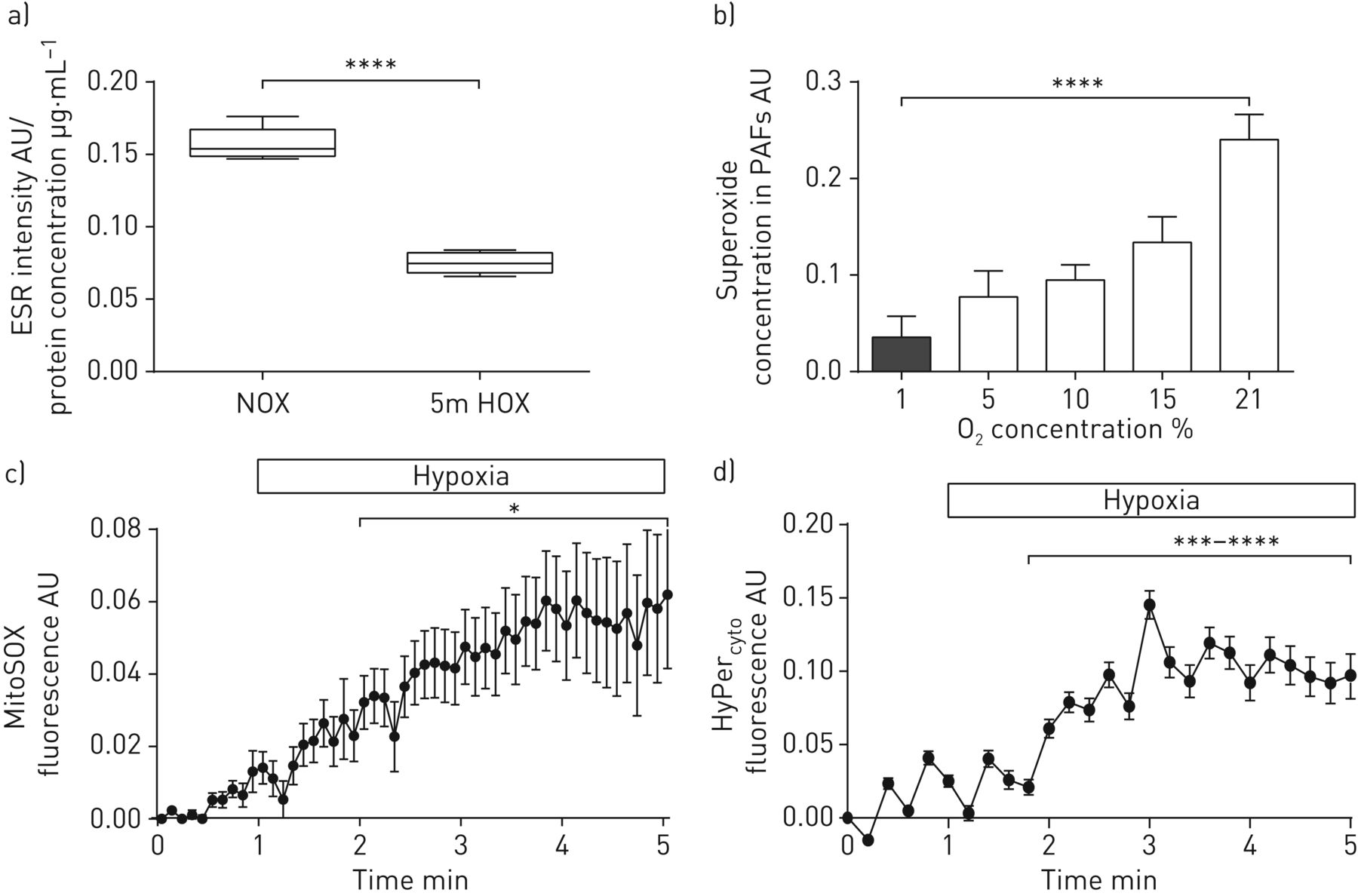
We further focused on superoxide release in the intact organ and the subcellular distribution of different ROS. In contrast to isolated PASMCs, the ROS/RNS levels in lung homogenate from intact lungs exposed to acute hypoxic ventilation were decreased as determined by the CMH probe (figure 2a). Accordingly, we detected decreased superoxide release from isolated lung fibroblasts during hypoxia (figure 2b). With regard to the intracellular distribution, acute hypoxia increased mitochondrial ROS in PASMCs measured by MitoSOX using fluorescence microscopy (figure 2c). The mitochondrial staining pattern of MitoSOX was confirmed by confocal microscopy (supplementary figure S3).

Effect of acute hypoxia on subcellular, cell-type-specific reactive oxygen species (ROS) concentration. a) Total intra- and extracellular ROS/reactive nitrogen species determined by 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine (CMH) intensity in lung homogenates from isolated ventilated and perfused lungs after 5 min of acute hypoxic (1% O2, 5.3% CO2, balanced with N2 (“5m HOX”)) or normoxic ventilation (21% O2, 5.3% CO2, balanced with N2 (“NOX”)). ESR: electron spin resonance; AU: arbitrary units. n=4 isolated lungs in each group. ****: p<0.0001 by the t-test with Welsh's correction. b) Superoxide concentration in primary mouse lung fibroblasts after 5 min of acute exposure to different O2 concentrations. The superoxide concentration was measured by ESR spectroscopy as the difference in the CMH signal with or without poly(ethylene glycol)-conjugated superoxide dismutase. PAF: pulmonary artery fibroblast. n=3–4 individual cell isolations per group. ****: p<0.0001 by one-way ANOVA with the Tukey post hoc test. c) Mitochondrial ROS concentration in pulmonary artery smooth muscle cells (PASMCs) during perfusion with hypoxic buffer. The level of mitochondrial ROS concentration is presented as the change in the MitoSOX fluorescent signal compared with baseline and normoxic values. n=4 individual cell isolations. *: p<0.05 by the t-test with Welsh's correction. d) Intracellular hydrogen peroxide (H2O2) concentration in PASMCs during perfusion with hypoxic buffer. The level of cellular H2O2 is presented as the change in the HyPercyto fluorescent signal compared with baseline and normoxic values. n=4 individual cell isolations. ***: p<0.001; ****: p<0.0001 by the t-test with Welsh's correction.
Given that H2O2 has been suggested as a downstream messenger during acute hypoxia [1], we next quantified cytosolic H2O2 by HyPercyto [25]. Acute hypoxia increased the concentration of H2O2 in PASMCs (figure 2d).
Effect of MitoQ on chronic hypoxia-induced pulmonary hypertension, PASMC proliferation and superoxide release in chronic hypoxia
MitoQ concentration after 4 weeks of treatment with 50 mg·kg−1·day−1 was in the range previously demonstrated to have protective effects in the heart (figure 3a) [26]. Exposure of mice to chronic hypoxia (10% O2, 4 weeks) induced an increase of the right ventricular systolic pressure (RVSP) to a similar degree in both the MitoQ- and TPP+-treated groups (figure 3b). In accordance, the degree of pulmonary vascular remodelling was increased after hypoxia, but not altered by MitoQ or TPP+ treatment (figure 3c). TPP+ treatment did not have any effect on RVSP or cardiac output during chronic hypoxia compared with untreated controls (supplementary figure S4).

Effect of MitoQ on development of chronic hypoxia-induced pulmonary hypertension (4 weeks, 10% O2). a) MitoQ concentration in mouse lung and liver after 4 weeks of treatment with MitoQ (60 mg·kg−1·day−1) via gavage measured by liquid chromatography and tandem mass spectrometry. b) Right ventricular systolic pressure (RVSP) and c) pulmonary vasculature remodelling in mice exposed for 4 weeks to chronic hypoxia (10% O2 (“4w HOX”)) or normoxia (21% O2 (“NOX”)) and treated with the mitochondria-targeted antioxidant (MitoQ) or the inactive carrier substance (triphenylphosphonium (TPP+)). n=8 animals per group. *: p<0.05; **: p<0.01; ****: p<0.0001 by two-way ANOVA with the Tukey post hoc test. d) Proliferation of pulmonary artery smooth muscle cells (PASMCs) after 5 days of hypoxic (1% O2 (“5d HOX”)) or normoxic (21% O2) incubation in the presence of MitoQ or TPP+. Proliferation is presented as percent of 5-ethynyluridine (EdU)-positive proliferating cells compared with the total cell number labelled by Hoechst staining. n=4 individual cell isolations. ***: p<0.001; ****: p<0.0001 by two-way ANOVA with the Tukey post hoc test. e) Superoxide concentration in PASMCs after 5 days of chronic hypoxic (1% O2) or normoxic (21% O2) incubation. The superoxide concentration was measured by electron spin resonance spectroscopy as the difference in the 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine signal with or without poly(ethylene glycol)-conjugated superoxide dismutase. AU: arbitrary units. n=4 individual cell isolations. ***: p<0.001 by the t-test with Welsh's correction. f) Mitochondrial ROS concentration determined by MitoSOX fluorescence in PASMCs after 5 days of chronic hypoxic (1% O2) or normoxic (21% O2) incubation. n=4 individual cell isolations. ****: p<0.001 by the t-test with Welsh's correction. g) Cytosolic H2O2 concentration determined by HyPercyto fluorescence in PASMCs after 5 days of hypoxic incubation (1% O2). n=4 individual cell isolations. *: p<0.05 by the t-test with Welsh's correction. h) Superoxide concentration per weight in lung homogenate from mice exposed to chronic hypoxia (10% O2) or normoxia (21% O2) for 4 weeks. n=3 individual cell isolations. i) Concentration of 8-hydroxyguanosine in DNA (diluted in buffer), cell lysate and growth medium from PASMCs exposed to chronic hypoxia (1% O2) or normoxia (21% O2) for 5 days. j) Superoxide concentration in PASMCs exposed to chronic hypoxia (1% O2) or normoxia (21% O2) for 5 days in the presence of 100 nM TPP+ or MitoQ. n=4 individual cell isolations. **: p<0.01 by two-way ANOVA with the Tukey post hoc test.
Moreover, low doses of MitoQ did not prevent hypoxia-induced proliferation of PASMCs, while higher doses showed an unspecific effect on proliferation, as TPP+ also decreased proliferation at high concentrations (figure 3d).
Total superoxide concentration, mitochondrial ROS levels and cellular H2O2 were decreased after 5 days of 1% O2 exposure of PASMCs measured by ESR spectroscopy, MitoSOX fluorescence and HyPercyto, respectively (figure 3e–g). Lung homogenate of mice after chronic hypoxia showed a tendency for decreased superoxide levels, but did not reach significance (figure 3h). All ROS measurements were performed in a continuous hypoxic environment, without reoxygenation (supplementary methods). Moreover, the level of 8-hydroxyguanosine, which is an indicator of DNA damage [27], was not changed in isolated DNA, PASMC lysates and in the medium after chronic hypoxic exposure of PASMCs compared with normoxic values (figure 3i). Antioxidative enzymes were upregulated at the mRNA level in PASMCs after chronic hypoxic exposure, but downregulated in lung homogenate of mice exposed to hypoxia, with the extracellularly located glutathione peroxidase 3 (GPX3) being the only enzyme upregulated at the mRNA and protein levels in PASMCs (supplementary figure S5a−d). Activity of SODs or catalase and total antioxidant capacity was, however, unchanged in chronic hypoxic PASMCs (supplementary figure S5e−g). The amount of mitochondrial DNA was decreased, while expression levels of enzymes indicating decreased mitochondrial glucose oxidation (pyruvate dehydrogenase kinase 1 (PDK1)) and increased anaerobic glycolysis (lactate dehydrogenase A (LDHA)) were increased (supplementary figure S6a−c). Inhibition of PDK1 by dichloroacetate could enhance mitochondrial superoxide concentration in normoxia, but not hypoxia (supplementary figure S6d).
At low concentrations, at which MitoQ did not inhibit proliferation of PASMCs (figure 3d), it decreased the superoxide concentration in normoxia compared with the normoxic untreated control (figure 3j). However, there were no differences in hypoxic superoxide concentration in untreated, TPP+-treated or MitoQ-treated PASMCs (figure 3j).
Effect of MitoQ on right ventricular remodelling in chronic hypoxia and after PAB
In contrast to the development of pulmonary hypertension/vascular remodelling, right ventricular remodelling differed after chronic hypoxia in the MitoQ- compared with the TPP+-treated group (figure 4). The ratio of the mass of the right ventricle to the mass of the left ventricle plus septum was significantly increased after hypoxic exposure in the TPP+- but not in the MitoQ-treated group (figure 4a). Echocardiographically determined right ventricular wall thickness (RVWT) was increased after chronic hypoxic exposure in both groups, albeit to a significantly lower level in the MitoQ group (figure 4c). Moreover, right ventricular internal diameter (RVID) and right ventricular outflow tract diameter (RVOTD) were not increased after chronic hypoxia in the MitoQ- in contrast to the TPP+-treated group, indicating less right ventricular dilatation during chronic hypoxia in the presence of MitoQ (figure 4c, d, h and i). Cardiac output and tricuspid annular plane systolic excursion (TAPSE) were decreased after chronic hypoxia, albeit to a similar degree in both the MitoQ- and TPP+-treated groups (figure 4e and f). Apart from these specific effects of MitoQ on the right ventricle, the carrier of MitoQ, TPP+, may have some additional effects on right ventricular remodelling, as RVWT and weight were decreased in the TPP+-treated group compared with the untreated group (supplementary figure S4).

Effect of MitoQ on remodelling of the right ventricle in mice exposed to chronic hypoxia for 4 weeks. a) Ratio of the mass of the right ventricle to the mass of left ventricle plus septum (RV/(LV+septum)) and echocardiographic parameters: b) right ventricular wall thickness (RVWT), c) right ventricular internal diameter (RVID), d) right ventricular outflow tract diameter (RVOTD), e) tricuspid annular plane systolic excursion (TAPSE) as the parameter for systolic right heart function and f) cardiac output (CO) in mice exposed to chronic hypoxia (10% O2 (“4w HOX”)) or normoxia (21% O2 (“NOX”)) for 4 weeks and treated with the mitochondria-targeted antioxidant (MitoQ) or the inactive carrier substance (triphenylphosphonium (TPP+)). n=8 animals per group. *: p<0.05; **: p<0.01; ***: p<0.001; ****: p<0.0001 and #: p<0.05; ##: p<0.01 interaction by two-way ANOVA with the Tukey post hoc test. g) Superoxide concentration in right ventricular homogenate in mice exposed to chronic hypoxia (10% O2) or normoxia (21% O2) for 4 weeks and treated with TPP+ or MitoQ. The superoxide concentration was measured by electron spin resonance spectroscopy as the difference in the 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine signal with or without poly(ethylene glycol)-conjugated superoxide dismutase. Data are presented as ratio to the organ mass. AU: arbitrary units. n=3–5. *: p<0.05 by two-way ANOVA with the Tukey post hoc test. h, i) Representative echocardiographic images of RVWT and RVOTD measurements in the right parasternal long axis: h) TPP+ and i) MitoQ. D: distance.
Furthermore, MitoQ application after PAB, which served as a hypoxia-independent stimulus for right ventricular hypertrophy, also improved right ventricular remodelling (figure 5). RVID was significantly reduced (figure 5c), while TAPSE was significantly increased (figure 5e) after PAB in MitoQ-treated mice compared with TPP+-treated mice.

Effect of MitoQ on remodelling of the right ventricle in mice after pulmonary arterial banding (PAB). a) Ratio of the mass of the right ventricle to the mass of the left ventricle plus septum (RV/(LV+septum)), b) right ventricular wall thickness (RVWT), c) right ventricular internal diameter (RVID), d) right ventricular outflow tract diameter (RVOTD), e) tricuspid annular plane systolic excursion (TAPSE), f) right ventricular systolic pressure (RVSP) and g) cardiac output (CO) in mice after PAB or sham operation. n=8 animals per group; **: p<0.01; ***: p<0.001; ****: p<0.0001 and #: p<0.05; ####: p<0.0001 interaction by two-way ANOVA with the Tukey post hoc test. h) Superoxide concentration in right ventricular homogenate in mice after sham or PAB operation and treated with triphenylphosphonium (TPP+) or MitoQ. Superoxide concentration was measured by electron spin resonance spectroscopy as the difference in the 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine signal with or without poly(ethylene glycol)-conjugated superoxide dismutase. Data are presented as ratio to the organ mass. AU: arbitrary units. n=5. *: p<0.05 and ##: p<0.01 interaction by two-way ANOVA with the Tukey post hoc test. i, j) Representative echocardiographic images of RVWT and RVOTD measurements in the right parasternal long axis: i) TPP+ and j) MitoQ. D: distance.
The superoxide concentration was significantly higher in right ventricular homogenate after chronic hypoxic exposure (supplementary figure S4h). MitoQ treatment decreased the superoxide concentration after chronic hypoxic exposure compared with the TPP+-treated group (figure 4g) and after PAB compared with the sham group (figure 5h).
Discussion
This study showed that increased superoxide, most likely originating from mitochondria, regulates acute, but not chronic hypoxia-induced pulmonary hypertension. This conclusion is based on the facts that 1) cellular superoxide and mitochondrial ROS production were increased after 5 min of hypoxic exposure of PASMCs and decreased after 5 days of hypoxic exposure, and 2) the mitochondria-targeted antioxidant MitoQ could inhibit the acute hypoxia-induced increase in superoxide, but not chronic hypoxia-induced pulmonary hypertension. Adaptation of mitochondrial metabolism/amount may underlie the differential role of ROS in acute and chronic hypoxia.
To the best of our knowledge, this is the first report investigating a specifically mitochondria-targeted antioxidant with regard to its effects on the response of the pulmonary vasculature to acute and chronic hypoxia.
Although there is recent evidence that mitochondria-derived superoxide plays an important role in acute and chronic hypoxic signalling in the pulmonary vasculature [3, 6, 28–30], two opposing concepts exist suggesting either an increase or decrease of ROS during hypoxia [3, 6, 28, 29]. This controversy is mainly based on the facts that 1) measurement of ROS during hypoxia is prone to artefacts, 2) localisation, time and species of ROS have to be taken into account, and 3) specific pharmacological antioxidants for mitochondria were not available. Here, we used three approaches to measure ROS: ESR technology, a fluorescent dye and a protein-based sensor. Moreover, the experiments in cells and tissue homogenates in this study were performed under continuous hypoxia, so that artefacts due to reoxygenation of the measurement sample were excluded. We identified that the concentration of mitochondrial ROS, as well as cytosolic H2O2, which acts as a possible downstream signalling mediator of superoxide [1], was increased in PASMCs during acute hypoxia. Mitochondrially produced superoxide can either be converted to H2O2 by mitochondrial SODs, which can diffuse into the cytosol or be converted to water by mitochondrially located GPXs, or be released from the mitochondria into the cytosol via voltage-dependent anion channels [31] where it can be converted to H2O2 by SOD1. Thus, either H2O2 or local superoxide (e.g. by colocalisation of mitochondria and ion channels) or both can act as downstream mediator(s) in acute hypoxia in PASMCs. Recently, we provided evidence that H2O2 originating from mitochondrial superoxide is essential for hypoxic signalling and HPV in PASMCs [30]. Although the mechanism for the superoxide release during acute hypoxia is still not fully elucidated, we could show that it depends on the presence of a specific isoform of the mitochondrial cytochrome c oxidase subunit 4 (Cox4i2) that causes mitochondrial hyperpolarisation in acute hypoxia, a condition well known to promote superoxide release from complex I or III of the mitochondrial respiratory chain [30]. Downstream targets for H2O2 are voltage-gated potassium channels [30], but possibly also several other ion channels [3, 32]. In contrast to PASMCs, acute hypoxia decreased superoxide concentration in pulmonary fibroblasts. This finding may explain why the ROS concentration in the lung homogenates was decreased during acute hypoxia, although our measurements could not discriminate between different ROS species in the whole lung. Nevertheless, our results could explain the discrepancy between measurements of ROS concentration in the whole lung and PASMCs by the fact of cell-type-specific regulation of ROS. Our findings are consistent with other studies showing increased mitochondrial ROS concentration in acute hypoxia [33–37]. The relevance of the increased superoxide in PASMCs for HPV was shown, as MitoQ inhibited the superoxide increase and HPV in our study.
The effect that inhibition of HPV reached a significant level at a slightly higher dose of MitoQ than the inhibition of hypoxia-induced superoxide release in PASMCs might be explained by the fact that isolated PASMCs may be more sensitive to MitoQ than PASMCs in isolated organs, where the substance has a higher distribution volume and diffusion distance.
MitoQ and TPP+ at high doses have been shown to inhibit oxidative phosphorylation [38] and interact with the mitochondrial sodium/calcium exchanger [39], thereby likely decreasing mitochondrial membrane potential. However, low doses of TPP+, which were used in this current study in vitro, did not alter mitochondrial respiration [40]. Thus, the effect of TPP+ on acute HPV and the hypoxia-induced increase in superoxide at concentrations >0.5 µM could be related to its property to decrease the mitochondrial membrane potential, as mitochondrial hyperpolarisation has been observed during acute HPV [37]. However, as MitoQ inhibited the acute hypoxic responses in the lower concentration range, while TPP+ did not, this effect can be regarded as specific for the antioxidant properties of MitoQ.
In addition to HPV, chronic hypoxia causes pulmonary hypertension due to pathological pulmonary vascular remodelling, which is characterised, in part, by proliferation of PASMCs of pulmonary vessels [18]. As in the case of HPV, evidence for both a decrease [41] and an increase of cellular ROS concentration triggering pulmonary hypertension development [8] has been provided. Recently, an elegant study showed that an increase of specifically mitochondrial H2O2 may promote development of chronic hypoxia-induced pulmonary hypertension [8]. In our study, the level of superoxide, mitochondrial ROS and cytosolic H2O2 measured by ESR spectroscopy, MitoSOX and HyPercyto fluorescence, respectively, was significantly decreased in the cytosol and mitochondria of PASMCs after 5 days of incubation in 1% O2 as well as in lung homogenate of chronically hypoxic mice, supporting the hypothesis that chronic hypoxia induces a decrease of total cellular ROS. Additionally, the level of 8-hydroxyguanosine (an indicator of oxidative-induced DNA damage [27]) was not altered during chronic hypoxia in PASMCs. Moreover, oral treatment with MitoQ reaching lung tissue levels that demonstrated protective effects in a previous study [26] did not inhibit chronic hypoxia-induced pulmonary hypertension or PASMC proliferation. Interestingly, we found downregulation of intracellular antioxidative enzymes but unchanged antioxidative capacity in chronic hypoxic PASMCs. Together with the finding of decreased mitochondrial glucose oxidation, indicated by decreased mitochondrial DNA amount, increased expression of PDK1 (which inhibits mitochondrial pyruvate oxidation) and increased expression of LDHA (which mediates anaerobic pyruvate metabolism), we suggest that adaptation of cellular metabolism causes the decreased ROS concentration in chronic hypoxia compared with acute hypoxia. In this regard, it was suggested previously that mitochondrial metabolism may be downregulated in chronic hypoxia to avoid excessive mitochondrial ROS production [42]. Interestingly, activation of mitochondrial glucose oxidation by dichloroacetate, which can inhibit hypoxia-induced pulmonary hypertension and human pulmonary arterial hypertension [43], could increase superoxide release in normoxia, but not hypoxia, indicating that mitochondrial adaptation to hypoxia-induced ROS levels is multifactorial and dichloroacetate inhibits pulmonary hypertension by a superoxide-independent mechanism. Thus, our data suggest that upregulation of mitochondrial ROS does not play a role in chronic hypoxia-induced pulmonary vascular remodelling due to diverse adaptive mechanisms that decrease superoxide concentration.
Moreover, decreased mitochondrial ROS in PASMCs do not seem to interact with proliferative pathways in PASMCs, as decreased superoxide levels in normoxia during MitoQ treatment also did not alter pulmonary vascular remodelling.
It cannot be excluded, however, that a cell-type-specific and localised subcellular release of specific ROS may activate proliferation pathways. Upregulation of other ROS sources might contribute to this finding, as hypoxia-induced ROS release might also originate from specific isoforms of the NADPH oxidase [4].
Interestingly, in chronic hypoxia, MitoQ application decreased the development of right ventricular hypertrophy and right ventricular dilatation. TPP+ also decreased right ventricular hypertrophy compared with untreated control animals, albeit to a lesser extent than MitoQ, and had almost no effect on right ventricular dilatation, suggesting that mitochondrial ROS specifically can influence right ventricular remodelling. Furthermore, MitoQ treatment attenuated the hypertrophy and dilatation of the right ventricle as well as prevented development of right ventricular dysfunction after PAB. Accordingly, MitoQ treatment decreased superoxide concentration in the right ventricle.
It has been shown previously that right ventricular remodelling may rely on ROS-dependent signalling pathways [5, 44], and that the transition from beneficial adaptive concentric right ventricular hypertrophy to right ventricular dilatation and failure may depend on the amount of ROS released [45]. Our study does not answer the question by which mechanisms mitochondrial ROS release is activated in the right ventricle, but suggests that increased right ventricular afterload and not hypoxia induces the increase in ROS. Moreover, the therapeutic potential and underlying mechanisms of MitoQ treatment on right ventricular dysfunction in pulmonary hypertension have to be further evaluated.
In conclusion, our study revealed that acute hypoxic exposure initiated an increase of mitochondrial ROS, most probably superoxide, in PASMCs, which regulates HPV, while chronic hypoxic exposure was associated with a decrease of the concentration of different ROS in PASMCs. Application of the mitochondria-targeted antioxidant MitoQ specifically attenuated HPV, but did not inhibit the development of pulmonary vascular remodelling and chronic hypoxia-induced pulmonary hypertension, while it attenuated right ventricular dilatation (figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of MitoQ in hypoxia. Acute hypoxia increases reactive oxygen species (ROS), while chronic hypoxia decreases ROS in the pulmonary vasculature, but increases ROS concentration in the right ventricle (RV). Mitochondria-targeted antioxidant MitoQ inhibits acute hypoxia-induced pulmonary vasoconstriction and chronic hypoxia, as well as pulmonary arterial banding (PAB)-induced right ventricular dilatation. LV: left ventricle; A: adventitia; M: media; E: endothelium; HPV: hypoxic pulmonary vasoconstriction.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary files ERJ-01024-2017_Supplement
Acknowledgements
The authors thank K. Homberger, I. Breitenborn-Mueller, N. Schupp, M. Wessendorf and E. Kappes (Excellence Cluster Cardio-Pulmonary System, Giessen, Germany) for technical assistance.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: None declared.
Support statement: Supported by the Deutsche Forschungsgemeinschaft AZ WE 1978/4-2 and SFB 1213, projects A06 and CP02, by the Medical Research Council UK (MC_U105663142) and by a Wellcome Trust Investigator award (110159/Z/15/Z) to M.P. Murphy.
- Received May 19, 2017.
- Accepted January 10, 2018.
- Copyright ©ERS 2018
References










