





Abstract
Pleural effusion is a frequent side-effect of dasatinib, a second-generation tyrosine kinase inhibitor used in the treatment of chronic myelogenous leukaemia. However, the underlying mechanisms remain unknown. We hypothesised that dasatinib alters endothelial integrity, resulting in increased pulmonary vascular endothelial permeability and pleural effusion.
To test this, we established the first animal model of dasatinib-related pleural effusion, by treating rats with a daily regimen of high doses of dasatinib (10 mg·kg−1·day−1 for 8 weeks).
Pleural ultrasonography revealed that rats chronically treated with dasatinib developed pleural effusion after 5 weeks. Consistent with these in vivo observations, dasatinib led to a rapid and reversible increase in paracellular permeability of human pulmonary endothelial cell monolayers as reflected by increased macromolecule passage, loss of vascular endothelial cadherin and zonula occludens-1 from cell–cell junctions, and the development of actin stress fibres. These results were replicated using human umbilical vein endothelial cells and confirmed by decreased endothelial resistance. Interestingly, we demonstrated that this increased endothelial permeability is a reactive oxygen species (ROS)-dependent mechanism in vitro and in vivo using a cotreatment with an antioxidant agent, N-acetylcysteine.
This study shows that dasatinib alters pulmonary endothelial permeability in a ROS-dependent manner in vitro and in vivo leading to pleural effusion.
Abstract
The drug dasatinib causes pleural effusion by disrupting the endothelial barrier with reactive oxygen species http://ow.ly/qOGE30gamGK
Introduction
Dasatinib is a potent second-generation tyrosine kinase inhibitor (TKI) approved for first-line treatment of Philadelphia-positive chronic myelogenous leukaemia (CML) and for all phases of Philadelphia-positive CML with resistance or intolerance to prior therapy, including imatinib. Up to around one-third of the patients (15–35%) treated with dasatinib develop pleural effusions, experiencing dyspnoea, chest pain, dry cough and decreased exercise tolerance. Fluid analyses reveal that these pleural effusions are generally lymphocyte-predominant exudates and could even be chylous [1–4].
Beyond this knowledge, the specific mechanisms underlying pleural fluid accumulation occurring during dasatinib therapy, and less commonly with other TKIs, are poorly defined. It has been described that a twice-daily dasatinib regimen, as well as the presence of hypertension, hypercholesterolaemia, skin rash, a history of cardiac or autoimmune diseases and the age of the patient, are strongly suspected as predisposing factors [1, 4–7]. However, our understanding of the molecular and cellular mechanisms is unclear, justifying the need for a reliable, clinically relevant animal model for dasatinib-related pleural effusion. Currently there is no animal model of nonmalignant pleural effusions, but only models of malignant pleural effusion that involve different underlying mechanisms [8].
Here, we tested the hypothesis that dasatinib alters endothelial integrity, which results in increased pulmonary vascular endothelial permeability and subsequently pleural effusion. Using clinically relevant doses of dasatinib chronically administered to Wistar rats, we obtained direct evidence establishing the connection between high doses of dasatinib and the development of pleural effusion [9]. Furthermore, we report here the first animal model of dasatinib-related pleural effusion that is similar to the phenomenon which occurs in humans.
Understanding the mechanisms by which dasatinib leads to pleural effusion will provide opportunities for intervention and for preventing this adverse event, and thus improve the management of nonmalignant pleural effusions.
Materials and methods
Additional details can be found in the supplementary material.
Animals and in vivo treatment
All animals were treated in accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the French National Institute of Health and Medical Research (INSERM) and approval was granted by the Ethics Committee of the University Paris-Sud (Le Plessis-Robinson, France). Young male Wistar rats (100 g; Janvier Labs, Saint Berthevin, France) were treated daily with a single intraperitoneal (i.p.) injection of dasatinib, imatinib or vehicle (dimethylsulfoxide (DMSO):saline (1:3, v/v)) for 8 weeks. Rats were anaesthetised with isoflurane via inhalation (2% in medical air). Thoracic and abdominal areas were shaved to perform transthoracic ultrasonography. Pleural micro-ultrasound (Vivid E9; GE Medical Systems, Milwaukee, WI, USA) was used to assess the presence of pleural fluid. Pulse-wave Doppler measurement of pulmonary outflow was recorded in the parasternal short-axis view at the level of the aortic valve to measure pulmonary artery velocity time integral and pulmonary valve acceleration time. There were no apparent effects of dasatinib at high dose on kidney or liver structures (data not shown).
Evaluation of lung permeability and lung wet/dry weight ratio
Animals were anaesthetised with isoflurane. Evans blue (30 mg·kg−1) was injected into the jugular vein 30 min before exsanguination. The blood was removed from the lungs. The right lung was homogenised in PBS (1 mL per 100 µg tissue), incubated with 2 volumes of formamide (18 h at 60°C) and centrifuged at 5000×g for 30 min. The optical density of the supernatant was determined spectrophotometrically at 620 and 740 nm using an EnVision plate reader (PerkinElmer, Courtaboeuf, France). The extravasated Evans blue concentration in the lung homogenate was calculated against a standard curve (micrograms of Evans blue dye per lung). The lung wet/dry weight (W/D) ratio was used as an index of lung water accumulation. To measure the total amount of lung water, the lung weight was measured immediately after its excision (wet weight). The lung tissue was then dried in an oven at 60°C for 5 days and reweighed as dry weight. The W/D ratio was calculated by dividing the wet weight by the dry weight.
Statistical analyses
Statistical significance was tested using the nonparametric Mann–Whitney U-test or two-way ANOVA with Bonferroni post hoc tests. Continuous data are expressed as mean±sem of at least three independent experiments or performed in triplicate or quintuplicate for technical replicates. A p-value <0.05 was considered statistically significant. Analyses were performed using Prism version 5.0 (GraphPad, La Jolla, CA, USA).
Results
Dasatinib dose-dependently causes pleural effusion in rats
To determine whether dasatinib could lead to pleural effusion, 4-week-old male Wistar rats, weighing 100 g, were divided into six groups and treated for 8 weeks with daily i.p. injections of: 1) vehicle, 2) dasatinib at 1 mg·kg−1·day−1, 3) dasatinib at 10 mg·kg−1·day−1, 4) imatinib at 1 mg·kg−1·day−1, 5) imatinib at 10 mg·kg−1·day−1 or 6) imatinib at 50 mg·kg−1·day−1 (supplementary figure S1). Based on a pharmacokinetic study performed in rats, we previously found that the dose of 1 mg·kg−1·day−1 of dasatinib was close to the clinically relevant dose (1×) [9].
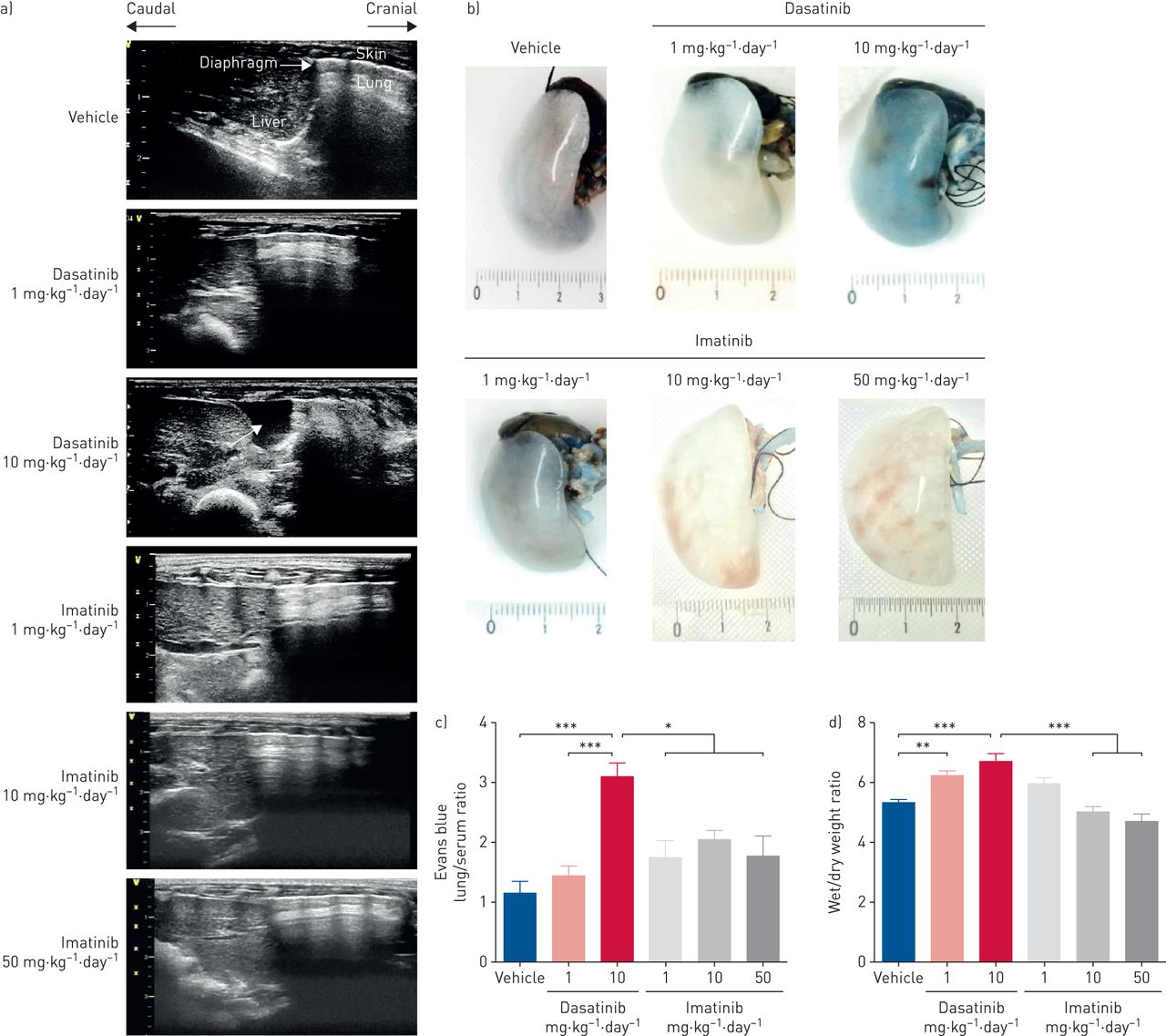
Using pleural ultrasonography, we found that rats treated with the high dose of dasatinib (10× the clinically relevant dose) developed pleural effusion over the period of 8 weeks, starting from week 5 (figure 1 and table 1). This was not observed in rats treated with dasatinib at the clinically relevant dose (1×) or rats treated with imatinib (figure 1 and table 1). Interestingly, no ascites and no pericardial effusion were observed in the rats (table 1). In addition, these observations support the notion that a dose–response relationship may exist, which mimicked the findings observed in humans [1, 7].

Chronic treatment of rats with high doses of dasatinib causes the development of pleural effusion. a) Representative images of pleural ultrasonographic images in all rats treated for 8 weeks with daily i.p. injections of vehicle (dimethylsulfoxide:saline (1:3, v/v)), dasatinib at 1 mg·kg−1·day−1 (clinical relevant dose (1×)) or 10 mg·kg−1·day−1 (10×), or imatinib at 1, 10 or 50 mg·kg−1·day−1. White arrow indicates pleural fluid. b) Representative images and c) quantification of vascular permeability assessed by Evans blue accumulation in the lung tissue from rats treated with vehicle, dasatinib or imatinib at the indicated concentrations. Ruler: cm. d) Quantification of the lung wet/dry weight ratio, used as an index of lung water, in rats treated with vehicle, dasatinib or imatinib at the indicated concentrations. c, d) Data presented as mean±sem (n=5–8). *: p<0.05; **: p<0.01; ***: p<0.001.
Incidence of pleural effusion, pericardial effusion and ascites over time, as determined by thoracic ultrasonography
Pleural effusion was paralleled by vascular leak, as substantial accumulation of Evans blue was observed in the lungs of rats treated with dasatinib (10×) when compared with the lungs of rats treated with vehicle, with dasatinib at the clinically relevant dose (1×) or with imatinib (figure 1b and c). In addition, the lung W/D ratio, used as an index of pulmonary oedema, was mildly elevated in lungs of dasatinib-treated rats 8 weeks after daily administration of dasatinib 1× or 10× when compared with vehicle-injected rats (figure 1d). No increase in Evans blue extravasation or W/D ratio was observed in imatinib-treated animals, suggesting a dasatinib-specific effect.
No significant changes were noted in the ejection fraction, stroke volume, heart rate and cardiac output in rats treated with high doses of dasatinib when compared with rats treated with vehicle or low doses of imatinib or dasatinib (table 2), suggesting that heart failure and increased filling pressures cannot explain the pleural effusion observed upon dasatinib exposure.
Cardiac parameters as measured by thoracic ultrasonography performed in the various treatment groups
Taken together, our results indicate that a high dose of dasatinib (10×) causes pleural effusion formation in rats, which closely mimicked the findings observed in humans. These in vivo observations indicate that dasatinib impairs endothelial barrier integrity.
Dasatinib increases endothelial cell permeability in a dose-dependent manner, a phenomenon that is reversible following TKI withdrawal
Maintaining endothelial barrier integrity is fundamental in the regulation of the passage of fluid, macromolecules and cells between the blood and the interstitial space. As loss of endothelial barrier integrity may contribute to pleural effusion [10–15], we next determined the impact of dasatinib on the integrity of endothelial cell monolayers in vitro. Endothelial permeability was evaluated using pulmonary endothelial cells derived from control lungs and using human umbilical vein endothelial cells (HUVECs), a well-established in vitro model of the post-capillary venule of the systemic circulation. Based on the pharmacokinetic profile of dasatinib [9, 16], pulmonary endothelial cells and HUVECs were exposed to increasing relevant doses of dasatinib: 10, 100 and 1000 nM were used as low, intermediate and high doses, respectively.
Human primary cultures of confluent pulmonary endothelial cell monolayers treated with dasatinib exhibited increased permeability to fluorescein isothiocyanate–dextran (40 kDa) compared with pulmonary endothelial cells treated with vehicle (figure 2a). The increase in macromolecule passage was paralleled by alterations in the distribution of tight junction molecule zonula occludens (ZO)-1 (figure 2b). In line with findings in human pulmonary endothelial cells, we found that dasatinib dose-dependently enhanced endothelial permeability in HUVECs, as evidenced by a dose-dependent drop in endothelial resistance (decrease in resistance by ∼20% and ∼30% at 100 and 1000 nM, respectively; p<0.01) (supplementary figure S2a and b). A dose-dependent increase in the passage of macromolecule solution (horseradish peroxidase, 60–70 kDa) paralleled the decrease in endothelial resistance (figure 2c). At the subcellular level, exposure of HUVEC monolayers to 100 nM dasatinib resulted in the formation intercellular gaps (figure 2d and supplementary figure S2c). As the intracellular domain of VE-cadherin directly binds to β-catenin that forms the link to α-catenin and the actin cytoskeleton, we next studied the β-catenin distribution by confocal microscopic analyses and immunolabelling with VE-cadherin in HUVECs treated with vehicle, dasatinib or imatinib. Consistent with the notion that dasatinib led to alterations in the distribution of key proteins for cell–cell junctions, we found a loss of β-catenin at the membrane in HUVECs treated with dasatinib when compared with vehicle or imatinib (supplementary figure S2c), indicating loss of adherens junction integrity. When exposing dasatinib-treated cells to thrombin, we found that dasatinib and thrombin had an additive effect, indicating that dasatinib exaggerates agonist-induced endothelial barrier disruption (supplementary figure S3).

High-dose dasatinib enhances endothelial permeability in cultured human pulmonary endothelial cells derived from control lungs and human umbilical vein endothelial cells (HUVECs). a) Quantification of macromolecule passage of fluorescein isothiocyanate–dextran over resting human pulmonary endothelial cells exposed for 3 h to vehicle (dimethylsulfoxide:saline (1:3, v/v): 0 nM) or increasing doses of dasatinib in the presence of 10% FCS. AU: arbitrary units. b) Confocal microscopic analyses and double labelling with antibodies directed against vascular endothelial (VE)-cadherin, zonula occludens (ZO)-1 together with 4′,6-diamidino-2-phenylindole (DAPI) in resting human pulmonary endothelial cells exposed for 3 h to vehicle or 100 nM dasatinib in the presence of 10% FCS. Arrows indicate interendothelial gaps. c) Quantification of macromolecule passage (horseradish peroxidase) over resting HUVEC monolayers after exposure to vehicle or dasatinib in 1% human serum albumin in M199 at different time-points. d) Confocal microscopic analyses and double labelling with VE-cadherin, F-actin together with DAPI in resting HUVECs exposed for 1 h to vehicle or 100 nM dasatinib. Arrow indicates interendothelial gap. a, c) Data presented as mean±sem (n=4–8). *: p<0.05 versus vehicle; #: p<0.05; ##: p<0.01 versus cells treated with 10 nM dasatinib; ¶: p<0.05 versus 100 nM dasatinib. b, d) Scale bar: 10 μm.
To exclude the possibility that dasatinib enhances endothelial permeability via increased cell death, we validated that dasatinib does not induce endothelial cell apoptosis over a period of 6 h under our culture conditions with complete media containing 10% FCS (supplementary figure S4). This was supported by the fact that the dasatinib-induced increase in endothelial permeability is reversible following TKI withdrawal in primary cultures of confluent pulmonary endothelial cell monolayers as reflected by the normalisation of the macromolecule passage (figure 3a) as well as reversal of the dasatinib-induced drop in endothelial resistance (figure 3b and c). Overall, dasatinib induces a dose-dependent increase in endothelial cell permeability in monolayers from both pulmonary and systemic origin, a phenomenon that is reversible following TKI withdrawal.

Dasatinib-induced increase in endothelial permeability is reversible after withdrawal. a) Quantification of macromolecule passage of fluorescein isothiocyanate–dextran over resting human pulmonary endothelial cells exposed to vehicle (dimethylsulfoxide:saline (1:3, v/v): 0 nM) or to dasatinib at the indicated concentrations in the presence of 10% FCS for 3 h before tyrosine kinase inhibitor withdrawal (24 h later). AU: arbitrary units. b) Electrical cell-substrate impedance sensing analysis of resting human umbilical vein endothelial cell (HUVEC) monolayers after incubation with dasatinib at the indicated concentrations (first arrow) and after dasatinib withdrawal (second arrow). c) Quantification of normalised resistance of HUVEC monolayers before addition of the indicated compounds (t=1 h), after addition of compounds (t=5 h) and after switch to normal culture conditions (t=8 h) (indicated by dashed lines in b). Data are presented as mean±sem (n=4–8). *: p<0.05; **: p<0.01 versus vehicle; ##: p<0.01 versus cells treated with dasatinib 10 nM.
Inhibition of the Src kinase Lyn or activation of the Rho kinase pathway does not explain the dasatinib-induced increase in endothelial permeability
As Lyn-targeting drugs, such as dasatinib, have the ability to alter endothelial barrier function [17] and Lyn was shown to be a critical factor in the maintenance of the endothelial barrier [15], we hypothesised that inhibition of Lyn kinase underlies the dasatinib-induced increases in endothelial permeability. In contrast to these studies, we found that silencing of Lyn using small interfering RNA (supplementary figure S5a and b) improved the endothelial barrier both in pulmonary endothelial cells (supplementary figure S5c and d) and in HUVECs (supplementary figure S5e), without affecting the dasatinib-induced increase in endothelial permeability (supplementary figure S5f).
Alternatively, dasatinib may disrupt the endothelial barrier by releasing a Src-driven brake on Rho kinase 2 (ROCK2) [18, 19], a kinase that drives actomyosin contraction. To evaluate whether ROCK mediates the effects of dasatinib, we first determined the phosphorylation status of myosin light chain (MLC) at Ser19, a signalling event which is central in endothelial actomyosin contractility [20, 21]. Compared with vehicle, dasatinib led to a dose-dependent increase in phosphorylated MLC (pMLC) Ser19 in human pulmonary endothelial cells (supplementary figure S6a) and in HUVECs (supplementary figure S6b).
To determine whether this ROCK activation contributes to the dasatinib-induced endothelial cell permeability, we next exposed confluent monolayers of HUVECs to the highly potent, cell-permeable, selective ROCK inhibitor Y-27632 at a high concentration of 10 μM, which was shown to completely inhibit ROCK [22]. Although Y-27632 abolished dasatinib-induced pMLC Ser19 levels and reversed the dasatinib-induced formation of stress fibres (supplementary figure S6b), no significant reduction in the passage of the macromolecule solution (horseradish peroxidase) was observed in the presence of Y-27632 (supplementary figure S6c).
Taken together, our results indicate that dasatinib harbours mechanisms other than Lyn inhibition or ROCK activation to induce barrier disruption of endothelial monolayers.
Dasatinib-induced increase in endothelial permeability is mediated through a reactive oxygen species-dependent mechanism
As dasatinib is known to reduce the cellular content of the major intracellular antioxidant defence glutathione (GSH) and to induce a marked increase in the level of reactive oxygen species (ROS) [9, 23–25], we hypothesised that dasatinib-induced endothelial cell permeability could be due to increased oxidative stress. ROS are involved in cell–cell junction disruption in both epithelial cells [26–28] and endothelial cells [13–15, 29], thereby leading to barrier dysfunction.
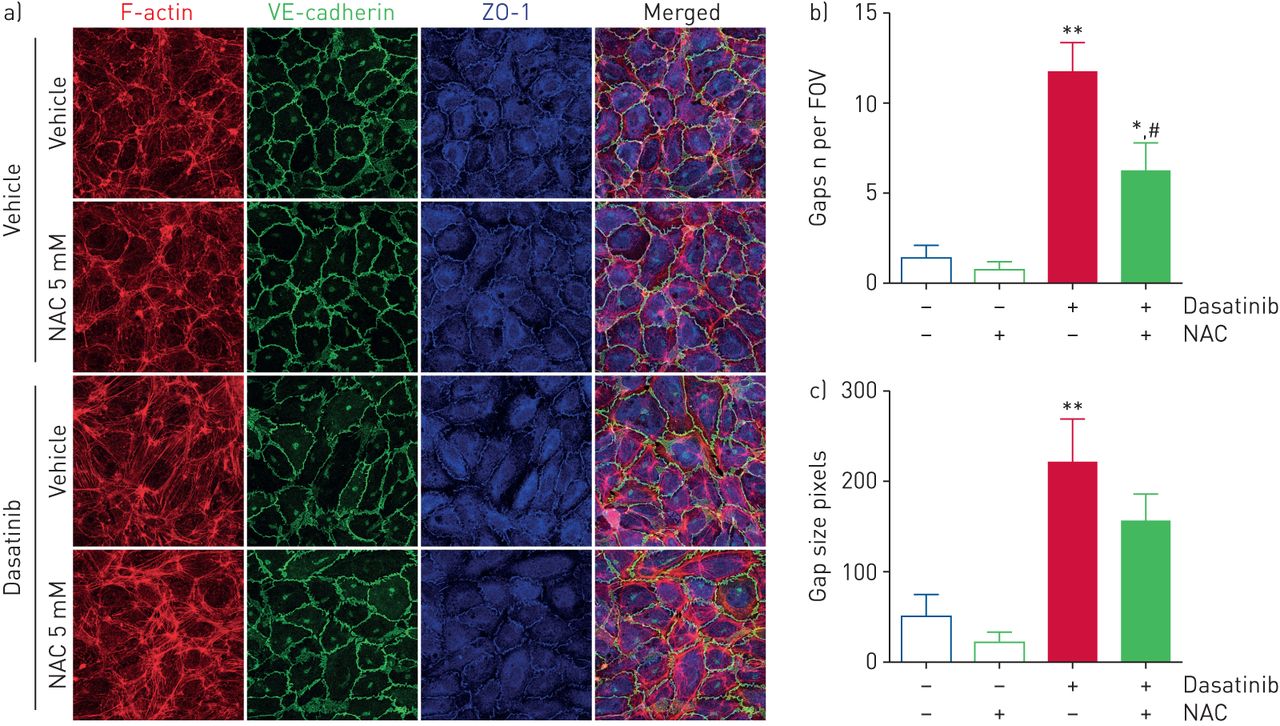
To test this hypothesis, we first determined the level of oxidative stress in rat lungs and found increased levels of lipid peroxidation in lung homogenates of rats treated with high doses of dasatinib when compared with lung homogenates of rats treated with low doses of dasatinib, vehicle or imatinib (figure 4a). Consistent with this observation, we also found a substantial increase in nitrotyrosine content in lungs of rats treated with high doses of dasatinib when compared with vehicle or high doses of imatinib, a phenomenon that was abolished in rats treated with the antioxidative agent N-acetylcysteine (NAC) (figure 4c, and supplementary figure S7a and b). Next, pulmonary endothelial cells and HUVECs were pre-treated with NAC at a dose of 5 mM before exposure to dasatinib. We found that NAC treatment partially attenuated alterations in the redistribution of VE-cadherin and ZO-1, and decreased macromolecule passage in human pulmonary endothelial cells (figure 4d and e). Although NAC reduced the number and size of endothelial gaps induced by 100 nM dasatinib in HUVECs (figure 5), no change in the passage of macromolecules (supplementary figure S7c) or the drop in endothelial resistance (supplementary figure S7d) was observed.
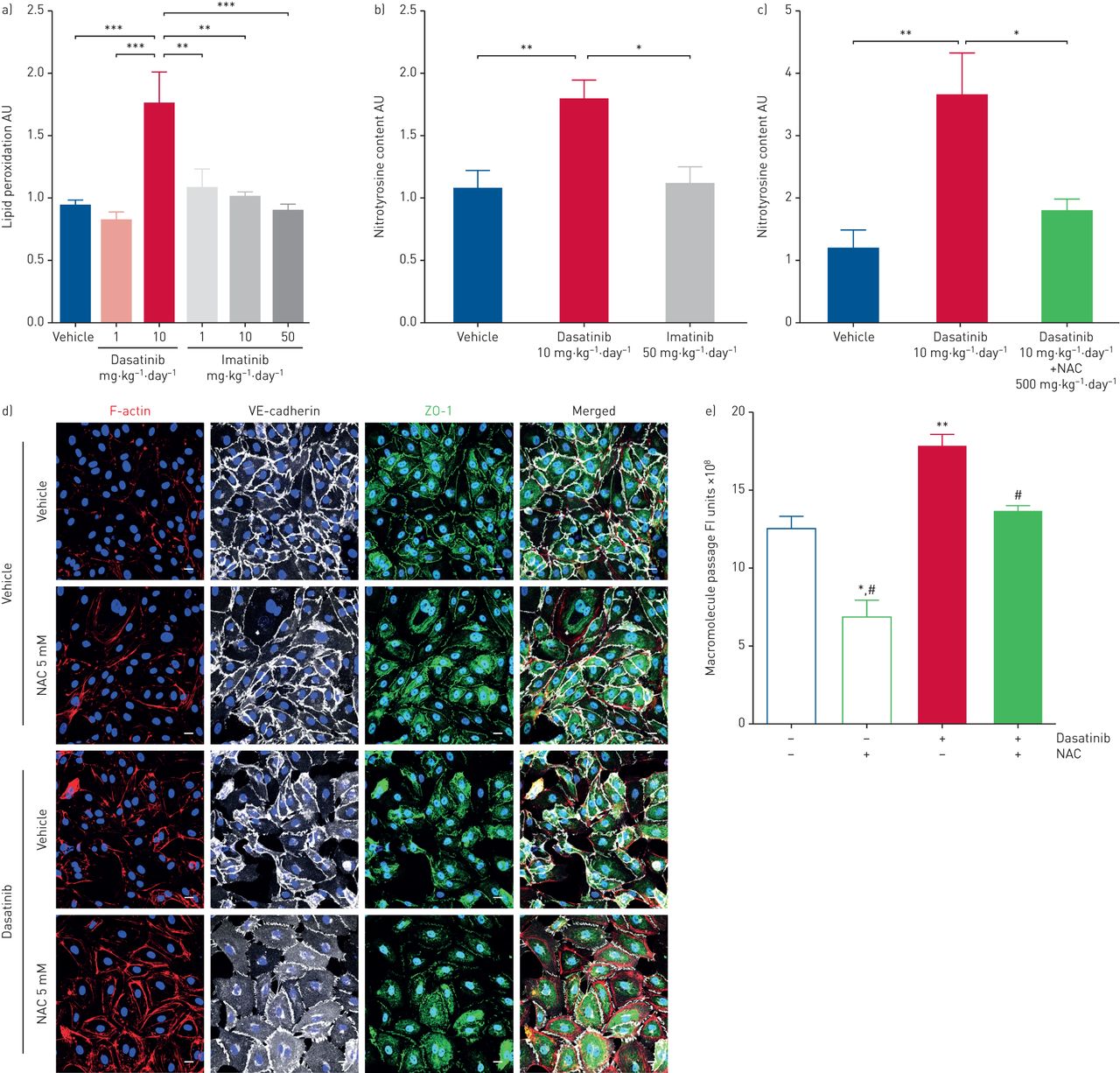
Dasatinib-induced increases in endothelial permeability is mediated through a reactive oxygen species-dependent mechanism. a) Quantification of lipid peroxidation levels in the lungs of rats treated with vehicle (dimethylsulfoxide:saline (1:3, v/v)), dasatinib or imatinib at the indicated concentrations. b) Quantification of nitrotyrosine content in the lungs of rats treated with vehicle, dasatinib at 10 mg·kg−1·day−1 or imatinib at 50 mg·kg−1·day−1 as determined by Western blot analyses (see supplementary material for full blots). AU: arbitrary units. c) Quantification of nitrotyrosine content in the lungs of rats treated with vehicle, dasatinib at 10 mg·kg−1·day−1 or dasatinib at 10 mg·kg−1·day−1 plus N-acetylcysteine (NAC) at 500 mg·kg−1·day−1 as determined by Western blot analyses (see supplementary material for full blots). d) Confocal microscopic analyses and double labelling of pulmonary endothelial cells with antibodies directed against F-actin, VE-cadherin, zonula occludens (ZO)-1 together with 4′,6-diamidino-2-phenylindole. Scale bar: 10 μm. e) Quantification of macromolecule passage of fluorescein isothiocyanate–dextran in resting human pulmonary endothelial cells pre-treated or not with the antioxidant agent NAC for 16 h and then exposed to vehicle or 1000 nM dasatinib in the presence of 10% FCS with or without NAC for an additional 3 h. FI: fluorescence intensity. a–c, e) Data are presented as mean±sem of n=3 donors (in vitro) or n=5–8 rats per group (in vivo). *: p<0.05; **: p<0.01; ***: p<0.001 versus vehicle or rats treated with dasatinib at 10 mg·kg−1·day−1; #: p<0.05 versus cells treated with 100 nM dasatinib.

Effect of the antioxidative agent N-acetylcysteine (NAC) on human umbilical vein endothelial cell (HUVEC) monolayer permeability. a) Confocal microscopic analyses and double labelling with antibodies directed against VE-cadherin and zonula occludens (ZO)-1, and direct labelling of F-actin (rhodamine phalloidin) and the nucleus (4′,6-diamidino-2-phenylindole). Scale bar: 10 μm. b, c) Quantification of the b) number and c) size of endothelial gaps induced by 100 nM dasatinib in the presence of 1% human serum albumin in M199 with or without NAC in HUVECs. FOV: field of view. Data are presented as mean±sem of n=2 donors with four images per donor. *: p<0.05; **: p<0.01 versus vehicle (dimethylsulfoxide:saline (1:3, v/v); #: p<0.05 versus cells treated with dasatinib.
The antioxidative agent NAC prevents dasatinib-induced pleural effusion in rats
Rats were chronically treated with dasatinib as described earlier, in the presence or absence of NAC at a dose of 500 mg·kg−1·day−1 (supplementary figure S7e). Pre-treatment with NAC abolished oxidative DNA modification in the lungs of rats, assessed by immunofluorescence for 8-oxo-2′-deoxyguanosine (8-oxo-dG) (figure 6a), and prevented dasatinib-induced pleural effusion in rats (figure 6b and table 3). Consistent with these observations, we found that pre-treatment with NAC prevented the accumulation of Evans blue in the lungs of rats treated with high doses of dasatinib (10×) and substantially decreased the W/D ratio (figure 6c and d).

The antioxidative agent N-acetylcysteine (NAC) prevents dasatinib-induced pleural effusion in rats. a) Confocal microscopic analyses and double labelling with antibodies against 8-oxo-2′-deoxyguanosine (8-oxo-dG) and Tie2 together with 4′,6-diamidino-2-phenylindole (DAPI) in lungs of rats chronically treated with vehicle (dimethylsulfoxide:saline (1:3, v/v)) or dasatinib at 10 mg·kg−1·day−1 with or without NAC at 500 mg·kg−1·day−1. Scale bar: 10 μm. b) Representative thoracic ultrasonographic images from rats treated with vehicle or dasatinib at 10 mg·kg−1·day−1 with or without NAC at 500 mg·kg−1·day−1. Arrow indicates pleural fluid. c) Quantification of lung vascular permeability as assessed by Evans blue accumulation and d) lung wet/dry weight ratio in rats treated with vehicle or dasatinib at 10 mg·kg−1·day−1 with or without NAC at 500 mg·kg−1·day−1. Data are presented as mean±sem (n=8). **: p<0.01; ***: p<0.001 versus rats treated with vehicle; #: p<0.05 versus rats treated with dasatinib at 10 mg·kg−1·day−1.
The effect of N-acetylcysteine (NAC) on the incidence of dasatinib-induced pleural effusion
No significant changes were noted in the ejection fraction, stroke volume, heart rate and cardiac output in rats treated with high doses of dasatinib in the presence or absence of NAC when compared with rats treated with vehicle (table 2).
These results support the notion that dasatinib-induced pleural effusion is mediated through a ROS-dependent mechanism.
Dasatinib affects pulmonary lymphatic structure and function
As pleural effusions are the product of an imbalance between endothelial barrier integrity, hydrostatic forces in the capillaries, and the capacity of lymphatics to collect the extravasated fluid and return it to the venous system, additional experiments were performed to determine whether dasatinib could alter lymphatic vessel structure and/or function. First, we studied the effect of dasatinib on lymphatic vessel structure in vivo by confocal microscopic analyses and double immunolabelling with lymphatic vessel hyaluronic acid endothelial receptor (Lyve)-1 and α-smooth muscle actin in lungs of rats treated with vehicle, dasatinib at 10 mg·kg−1·day−1 or imatinib at 50 mg·kg−1·day−1. Interestingly, we found a dilatation of lymphatic vessels in lungs of rats treated with high doses of dasatinib when compared with rats treated with vehicle or high doses of imatinib (figure 7a). In addition, we studied the in vitro effect of dasatinib on the electrical resistance of human pulmonary lymphatic endothelial cell (HPLEC) monolayers. Consistent with our observations obtained in human pulmonary endothelial cells and HUVECs, we found that dasatinib enhanced endothelial permeability in confluent HPLEC monolayers, as evidenced by a substantial, but reversible drop in endothelial resistance (decrease in resistance by ∼35%; p<0.001) (figure 7b and c, and supplementary figure S8a), and alterations in the distribution of VE-cadherin and β-catenin in HPLECs treated with 100 nM dasatinib when compared with vehicle (supplementary figure S8b).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dasatinib affects pulmonary lymphatic structure and function. a) Immunofluorescence staining of lymphatic vessels (arrows) in lung of rats treated with vehicle (dimethylsulfoxide:saline (1:3, v/v)), dasatinib at 10 mg·kg−1·day−1 or imatinib at 50 mg·kg−1·day−1. α-Smooth muscle actin (α-SMA) is used as the smooth muscle marker (red); lymphatic vessel hyaluronic acid endothelial receptor (Lyve)-1 is used as the marker for lymphatic vessels (green). Scale bar: 50 μm. b, c) Electrical cell-substrate impedance sensing analysis of resting human pulmonary lymphatic endothelial cell monolayers during treatment with 100 nM dasatinib or vehicle: b) longitudinal measurement of absolute resistance and c) drop in resistance 1 h after stimulation with 100 nM dasatinib (dashed line in b). Data are presented as mean±sem of n=7–8 measurements in two independent experiments. ***: p<0.001 in unpaired t-test.
Discussion
Pleural effusions are a frequent complication of dasatinib use. However, the underlying mechanisms remain unknown because of the lack of an animal model of nonmalignant pleural effusions. Here, we established the first animal model of dasatinib-related pleural effusion, by treating rats with a daily regimen of high doses of dasatinib (10× clinical relevant dose for 8 weeks). Interestingly, our present report demonstrates that rats with a daily regimen of high doses of dasatinib have an altered endothelial barrier integrity, which results in increased pulmonary vascular endothelial permeability leading to pleural effusion. We performed in vitro studies using human primary cultures of pulmonary endothelial cells derived from human lung tissues, HUVECs and HPLECs. We found that dasatinib increases in vivo and in vitro endothelial permeability in a dose-dependent fashion, through production of ROS. This phenomenon is reversible following dasatinib withdrawal and is not sufficiently explained by ROCK activation or Lyn inhibition. We also showed that dasatinib leads to alterations in the distribution of key proteins for cell–cell junctions, such as VE-cadherin and ZO-1, and to the development of actin stress fibres. Cotreatment with the antioxidant agent NAC in vitro and in vivo prevents the dasatinib-induced increase in endothelial permeability and the development of pleural effusion, indicating that ROS production underlies dasatinib-induced endothelial barrier disruption.
It is well known that pleural effusion is a frequent side-effect of dasatinib that is even more prominent if high doses and/or a twice-daily dosing schedule are used, particularly in advanced disease [1, 7]. Although dasatinib was shown to increase endothelial barrier permeability [30], pleural effusion has not been evaluated and dose dependency was not shown. Our understanding of the underlying mechanisms remains limited due to the lack of a reliable, clinically relevant animal model accurately reflecting dasatinib-induced pleural effusion. Other TKIs, such as imatinib, nilotinib and bosutinib, are associated with low incidences of pleural effusion [1, 4, 31–34], while some have even been associated with enhancement of the endothelial barrier [35, 36]. Further studies are therefore needed to identify which potential protein tyrosine kinases targeted by dasatinib are specifically involved in pleural effusion formation. A comparison of inhibition profiles of these specific TKIs with dasatinib across the whole kinase panel could help to reveal these contributing dasatinib target(s), and thus could facilitate the design of new drugs and also improve our understanding of the underlying mechanisms of pleural fluid accumulation.
Here, we obtained direct evidence establishing a connection between high doses of dasatinib and the development of pleural effusion. Interestingly, our results support the notion that a dose–response relationship may exist, which mimicked the findings observed in humans [1, 7]. Indeed, only rats treated with 10× clinical doses of dasatinib developed pleural effusion following a period of at least 5 weeks. To unravel the mechanism(s) responsible for the dasatinib-induced pleural effusion, we performed in vitro studies using HUVECs, HPLECs and human pulmonary endothelial cells exposed or not to increasing doses of dasatinib. Consistent with our in vivo observations, our findings show that dasatinib favours endothelial permeability in vitro in a dose-dependent and reversible manner. In all experiments performed in our study, a 100 nM concentration was sufficient to induce barrier disruption, a concentration that corresponds well to levels found in some CML patients. This may indicate that the individual patient's pharmacokinetics of dasatinib determine the risk for developing pleural effusion as a side-effect of dasatinib. As the integrity of interendothelial junctions consisting of tight junctions and adherens junctions can be impaired by endothelial cell retraction and shape change, we performed a series of experiments with the aim of determining whether dasatinib can cause intercellular endothelial gap formation. We found that dasatinib led to the formation of cytoplasmic stress fibres as well as loss of VE-cadherin, β-catenin and ZO-1 from intercellular junctions.
As dasatinib and its metabolites are known to covalently bind GSH [23, 24], and to induce a marked increase in ROS production and reduce the cellular content in GSH [9, 23–25], we focused our attention on the importance of the role of the oxidative stress. Indeed, ROS are involved in cell–cell junction disruption in both epithelial cells [26–28] and endothelial cells [13–15, 29]. Consistent with this notion, we recently demonstrated that chronic dasatinib treatment leads to a dose-dependent increase in mitochondrial ROS production that contributes to pulmonary endothelial dysfunction and predisposes to the development of pulmonary hypertension [9]. Our present results clearly indicate that attenuation of dasatinib ROS production by NAC can prevent the increased pulmonary endothelial permeability in vitro and the development of pleural effusion in rats. These observations are consistent with the fact that ROS, including superoxide and hydrogen peroxide, could alter the function of several proteins, including junctional proteins, via protein modification such as thiol oxidation, phosphorylation, nitration and carbonylation [37–40]. Consistent with these notions, Rao et al. [37] reported that oxidative stress induces tyrosine phosphorylation and cellular redistribution of occludin/ZO-1 and E-cadherin/β-catenin complexes by a tyrosine kinase-dependent mechanism. A similar phenomenon was observed in cultured endothelial cells exposed to high doses of dasatinib. These findings are consistent with the fact that none of the rats cotreated with NAC and high doses of dasatinib (10×) developed pleural effusion. A question that shapes future research is whether the effects of dasatinib result from the inhibition of one or more of the dasatinib-sensitive kinases, or whether a certain threshold concentration triggers ROS production in a specific toxic manner, in particular because the main dasatinib-sensitive kinases (Abl and Src kinases) mediate the permeability response. The finding that very low dasatinib concentrations (<10 nM) protect the endothelial barrier is in line with this thought. The role of Lyn in endothelial barrier regulation remains unclear, as Lyn was shown to strengthen the endothelial barrier [17, 41], while our experiments demonstrated a slight improvement in endothelial barrier function upon Lyn depletion (supplementary figure S5).
As the signal transduction pathways that cause intercellular endothelial gap formation involve a complex series of signalling events leading ultimately to rapid and sustained phosphorylation of MLC [42, 43], we also studied the phosphorylation status of MLC and tested the efficacy of a specific ROCK inhibitor Y-27632. Although our results showed increased ROCK activation in our cultured endothelial cells exposed to dasatinib, no beneficial effect of Y-27632 was noted. Similarly, we found that the inhibition of the Src family protein tyrosine kinase Lyn by a small interfering RNA approach in pulmonary endothelial cells was not implicated in the dasatinib-induced increase in endothelial permeability in vitro. As there is variability among cells freshly isolated from each patient, we cannot exclude the possibility that in vivo RhoA/ROCK activation or Lyn inhibition may also partly contribute to the dasatinib-induced increase in endothelial permeability. Consistent with this notion, Dasgupta et al. [30] recently reported that in vivo chronic administration of Y-27632 significantly inhibited the dasatinib-induced extravasation of Evans blue in mice and that the dasatinib-induced increase in microvascular permeability was attenuated in ROCK1-deficient mice.
Although our work indicates that pulmonary endothelium is more susceptible to dasatinib toxicity, further studies are needed to dissect the underlying mechanisms. Indeed, our in vitro results showed that dasatinib induces approximately two-fold endothelial gap formation in human pulmonary endothelial cells versus HUVECs. In addition, no pericardial effusion or ascites were observed in our rats treated with high doses of dasatinib, which is in contrast to the fact that dasatinib-treated CML patients are at risk for the development of pericardial effusions [1, 7]. A potential explanation for the lung specificity of dasatinib effects could be related to the high transcapillary hydrostatic gradient in lungs (due to the low interstitial pressure), and in pleural capillaries and venules (due to the negative intrathoracic pressure), which favours movement of fluid from the pulmonary capillaries into the interstitium and alveolar spaces. In contrast, it is known that the interstitial pressure is much lower in the pericardium and in the abdomen. Although we have been unable to collect pleural fluids without blood contamination for the biological assessment of these pleural fluids in rats treated with high doses of dasatinib due to their small size, we interestingly obtained data supporting that dasatinib affects the pulmonary lymphatic structure and monolayer permeability. Indeed, we found a substantial dilatation of lymphatic vessels rats treated with high doses of dasatinib, and that dasatinib can enhance the in vitro endothelial permeability in confluent HPLEC monolayers and alter the distribution of VE-cadherin and β-catenin. It remains to be determined, however, how these lymphatic changes affect the draining capacity of the lymphatic vessels. Therefore, further studies are needed to better understand the effect of dasatinib on pleural microcirculation as well as pulmonary lymphatic structure and function, including a better evaluation of pleural fluids in this animal model of dasatinib-related pleural effusion.
In summary, our work highlights that high circulating levels of dasatinib alter pulmonary endothelial permeability in a ROS-dependent manner in vitro and in vivo, leading to pleural effusion. Even if lowering the dasatinib dose or shifting to another TKI, when it is possible, are the best current options in the clinic, a better understanding of the underlying mechanisms of pleural effusion may help in the management of patients treated with dasatinib with recurrent or chronic pleural effusions. This first animal model of dasatinib-related pleural effusion will certainly help to understand the underlying mechanisms by which dasatinib leads to pleural effusion and to ameliorate the management of nonmalignant pleural effusions.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary materials and methods, and supplementary figures ERJ-01096-2017_Supplementary_material
Disclosures
Supplementary Material
M. Humbert ERJ-01096-2017_Humbert
D. Montani ERJ-01096-2017_Montani
A. Seferian ERJ-01096-2017_Seferian
A. Vonk Noordegraaf ERJ-01096-2017_Vonk_Noordegraaf
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: This research was supported by grants from the French National Institute for Health and Medical Research (INSERM), the University Paris-Sud and University Paris-Saclay, the Marie Lannelongue Hospital, the National Agency for Drug Safety (ANSM) grant VIGIAPATH, and in part by the Dépt Hospitalo-Universitaire (DHU) Thorax Innovation (TORINO), the Assistance Publique-Hôpitaux de Paris (AP-HP), Service de Pneumologie, Centre de Référence de l'Hypertension Pulmonaire Sévère, the LabEx LERMIT (grant ANR-10-LABX-0033), and the French Pulmonary Hypertension Patient Association (HTAP France). C. Phan was supported by the French Fonds de Dotation “Recherche en Santé Respiratoire” (FRSR) Fondation du Souffle (FdS). E.M. Jutant was the recipient of an Année-Recherche fellowship from AP-HP. J. Aman was funded by the Dutch Heart Foundation (Nederlandse Hartstichting; grant 2014T064). Funding information for this article has been deposited with the Crossref Funder Registry.
Conflict of interest: Disclosures can be found alongside this article at erj.ersjournals.com
- Received May 31, 2017.
- Accepted October 14, 2017.
- Copyright ©ERS 2018
References










